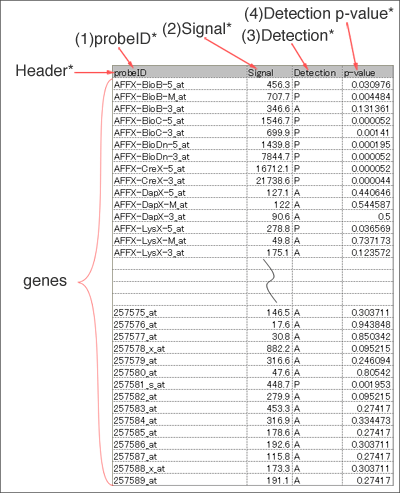
Data format for microarray data
For the platform "AG" and "ATH1", calculate signals using MAS5 (sample code 1).
Input file should include header line and following four fields (1~4). Fill all the blanks with "NA" (blank is not allowed). Save the table as Microsoft Excel (.xls, .xlsx) or tab-delimited text file.
*If you want to analyze microarray from platform "Arabidopsis Gene 1.1 ST Array", calculate signals using RMA (sample code 2). In this case, input files should contain (1) and (2).
For the other platforms, use AGI code (Locus id i.e. At1g00100) instead of probe ID.
You can get a sample file by clicking on this picture.
*If you want to analyze microarray from platform "Arabidopsis Gene 1.1 ST Array", calculate signals using RMA (sample code 2). In this case, input files should contain (1) and (2).
For the other platforms, use AGI code (Locus id i.e. At1g00100) instead of probe ID.
You can get a sample file by clicking on this picture.
Sample code 1 (R): Calculation of MAS5-signals from cel files
Install package "affy" in R. Execute following code. Input files for AtCAST will be generated in the same directory to your cel files.setwd("where_your_cel_files_are") #Modify this line
library(affy)
data <- ReadAffy()
data.signals <- mas5(data)
data.mas5calls = mas5calls(data)
data.mas5pvals <- assayData(data.mas5calls)[["se.exprs"]]
data.file.list <- dir()
n = 0
a <- as.list(NULL)
for(data.file in data.file.list) {
if(any(i <- grep("CEL|cel", data.file))){
n = n+1
a[n] <- gsub("\\.CEL.gz|\\.cel.gz|\\.CEL|\\.cel", "\\.txt", data.file)
sa <- exprs(data.signals)[,n]
sb <- exprs(data.mas5calls)[,n]
sc <- data.mas5pvals[,n]
sd <- cbind(sa, sb, sc)
filename <- as.character(a[n])
x <- c("ProbeSetName", "Signal", "Detection", "p-value")
write(x, file=filename, sep="\t", ncolumns = 4)
write.table(sd, file=filename, append = TRUE, quote = FALSE, sep="\t", col.names = FALSE)
}
}
Sample code 2: Calculation of RMA-signals from cel files for Arabidopsis Gene 1.1 ST Array
Install packages "oligo" and "pd.aragene.1.1.st" in R. Execute following code. Input files for AtCAST will be generated in the same directory to your cel files.setwd("where_your_cell_files_are") #Modify this line
library(oligo)
geneCELs <- list.celfiles(full.names = TRUE)
affyGeneFS <- read.celfiles(geneCELs)
geneCore <- rma(affyGeneFS, target = "core")
featureData(geneCore) <- getNetAffx(geneCore, "transcript")
data.list <- dir()
n = 0
a <- as.list(NULL)
for(data.file in data.list) {
if(any(i <- grep("CEL|cel", data.file))){
n = n+1
a[n] <- gsub("\\.CEL|\\.cel|\\.CEL.gz|\\.cel.gz", "\\_r.txt", data.file)
sa <- exprs(geneCore)[,n]
filename <- as.character(a[n])
x <- c("ProbeSetName", "Signal")
write(x, file=filename, sep="\t", ncolumns = 2)
write.table(sa, file=filename, append = TRUE, quote = FALSE, sep="\t", col.names = FALSE)
}
}