Data format for RNA-seq data
Calculate gene counts (Tutorial).
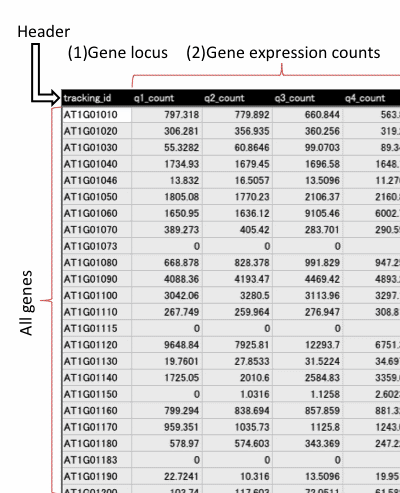
Input file should include header line and more than 3 fields (1~2). Fill all the blanks with "NA" (blank is not allowed). Save the table as Microsoft Excel (.xls, .xlsx) or tab-delimited text file.
You can get a sample file by clicking on this picture.
You can get a sample file by clicking on this picture.
Tutorial1: Calculation of Gene counts using Subread
1. Install R.2. Copy and paste following commands to R console.
2.1. Installation of Rsubread
x <- library(character.only=T)
if(length(grep("^Rsubread", x$results[,1], value=T))==0){ #Checking installation of Rsubread
source("http://bioconductor.org/biocLite.R")
biocLite("Rsubread")
}
if(length(grep("^RCurl", x$results[,1], value=T))==0){ #Checking installation of RCurl
source("http://bioconductor.org/biocLite.R")
biocLite("RCurl")
}
library(RCurl)
url <- "ftp://ussd-ftp.illumina.com/Arabidopsis_thaliana/Ensembl/TAIR10/Arabidopsis_thaliana_Ensembl_TAIR10.tar.gz"
userpwd <- "igenome:G3nom3s4u"
bin = getBinaryURL(url, userpwd = userpwd,
ftp.use.epsv = FALSE)
writeBin(bin, "Arabidopsis_thaliana_Ensembl_TAIR10.tar.gz")
untar("Arabidopsis_thaliana_Ensembl_TAIR10.tar.gz")
library(Rsubread)
buildindex(basename="arabidopsis_index", reference="./Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/WholeGenomeFasta/genome.fa")
If you have your own read data, DO NOT USE this command. Command "getwd()" in R, copy your fastq or fastq.gz files to a directory returned by the command.
##SRAdb install
if(length(grep("^SRAdb", x$results[,1], value=T))==0){ #Checking installation of SRAdb
source("http://bioconductor.org/biocLite.R")
biocLite("SRAdb")
}
##Download fastq files (in SRA project SRP003951 for example)
library(SRAdb)
sqlfile <- "SRAmetadb.sqlite"
if(!file.exists("SRAmetadb.sqlite")){ sqlfile <- getSRAdbFile() }
sra_con <- dbConnect(SQLite(),sqlfile)
rs = getFASTQinfo ( c("SRP003951"), srcType = 'ftp', sra_con = sra_con ) # Replace this line
getSRAfile( rs$run, sra_con, fileType = 'fastq')
library(Rsubread)
#List fastq files
fastq.files <- list.files(".", pattern="fastq")
paired.fastq.files.1 <- fastq.files[grep("_1.",fastq.files)]
paired.fastq.files.2 <- fastq.files[grep("_2.",fastq.files)]
single.fastq.files <- fastq.files[grep("_1.|_2.",fastq.files, invert=T)]
##Mapping of single-end reads
for(fastq.file in single.fastq.files){
output.file <- sprintf("%s_subread_results.bam", strsplit(fastq.file, ".fastq")[[1]][1])
align(index="arabidopsis_index", readfile1=fastq.file, output_file=output.file)
}
##Mapping of paired-end reads
if(length(paired.fastq.files.1)==length(paired.fastq.files.2)){
num.pairs <- length(paired.fastq.files.1)
for(j in 1:num.pairs){
output.file <- sprintf("%s_subread_results.bam", strsplit(paired.fastq.files.1[j], "_1.")[[1]][1])
align(index="arabidopsis_index", readfile1=paired.fastq.files.1[j], readfile2=paired.fastq.files.2[j], output_file=output.file, nthreads=3)
}
}else{
cat("Check that all fastq files are paired\n")
}
Read counts will be recorded in "counts.txt". Command "getwd()" in R to get where the file is. Input this file to AtCAST.
##single-end reads
input.files <- NULL
if(length(single.fastq.files) > 0){
for(i in 1:length(single.fastq.files)){
input.files[i] <- sprintf("%s_subread_results.bam", strsplit(single.fastq.files[i], ".fastq")[[1]][1])
}
list <- featureCounts(files=input.files, annot.ext="./Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf", isGTFAnnotationFile=TRUE)
}
##paied-end reads
if(length(paired.fastq.files.1) > 0 && length(paired.fastq.files.1)==length(paired.fastq.files.2)){
num.pairs <- length(paired.fastq.files.1)
for(j in 1:num.pairs){
input.files[j] <- sprintf("%s_subread_results.bam", strsplit(paired.fastq.files.1[j], "_1.")[[1]][1])
}
list <- featureCounts(files=input.files, annot.ext="./Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf", isPairedEnd=TRUE, isGTFAnnotationFile=TRUE)
}else{
cat("Check that all fastq files are paired\n")
}
write.table(list, file = "counts.txt", sep = "\t")
Tutorial2: Calculation of Gene counts using cufflinks
1. Install boost, sra_sdk, bowtie2, sam-tools, tophat2 and cufflinks following instructions of tophat2 and cufflinks.2. Download Arabidopsis ENSEMBL TAIR10 genome and annotation files (compressed in a file) from http://tophat.cbcb.umd.edu/igenomes.shtml, and decompress in a working folder.
3. Prepare your RNA-seq data. (Example we use: NGS seq files (SRR520237.sra, SRR520238.sra, SRR520239.sra, SRR520246.sra, SRR520247.sra, SRR520248.sra) were downloaded from SRA(NCBI) in the same folder and renamed (wt1_SRR520237.sra, wt2_SRR520238.sra, wt3_SRR520239.sra, pifq1_SRR520246.sra, pifq2_SRR520247.sra, pifq3_SRR520248.sra)).
4. Map sequences to Arabidopsis genome. (Example: Execute following commands. Length of reads in the example files were 50 bp.)
fastq-dump wt1_SRR520237.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_wt_rep1 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome wt1_SRR520237.fastq &&
fastq-dump pifq1_SRR520246.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_pifq_rep1 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome pifq1_SRR520246.fastq &
fastq-dump wt2_SRR520238.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_wt_rep2 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome wt2_SRR520238.fastq &&
fastq-dump pifq2_SRR520247.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_pifq_rep2 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome pifq2_SRR520247.fastq &
fastq-dump wt3_SRR520239.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_wt_rep3 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome wt3_SRR520239.fastq &&
fastq-dump pifq3_SRR520248.sra && # This line is not necessary if you use fastq file of your own.
tophat -r 50 -G ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Archives/archive-2011-08-30-20-11-05/Genes/genes.gtf -o tophat_pifq_rep3 ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Sequence/Bowtie2Index/genome pifq3_SRR520248.fastq &
cuffdiff ./Arabidopsis_thaliana_Ensembl_TAIR10/Arabidopsis_thaliana/Ensembl/TAIR10/Annotation/Genes/genes.gtf tophat_wt_rep1/accepted_hits.bam tophat_wt_rep2/accepted_hits.bam tophat_wt_rep3/accepted_hits.bam tophat_pifq_rep1/accepted_hits.bam tophat_pifq_rep2/accepted_hits.bam tophat_pifq_rep3/accepted_hits.bam &&